

一. PCR的发展历史
PCR技术自问世以来,在遗传病、病原体、癌基因等分子诊断领域和法医鉴定等方面发挥了巨大作用。代 PCR在进行扩增后通过凝胶电泳进行定性分析。
随着生物分子荧光技术的发展,1992年实时荧光定量PCR(Quantitative Real-time PCR, qPCR) 应运而生。qPCR是一种在DNA扩增反应中,以荧光化学物质荧光强度来测定每次聚合酶链式反应(PCR)循环后产物总量的方法。由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以Ct值也就成为定量的依据。基于荧光探针或染料的第二代 PCR 技术随后逐渐发展为检测核酸目标片段的主流分子诊断学技术。
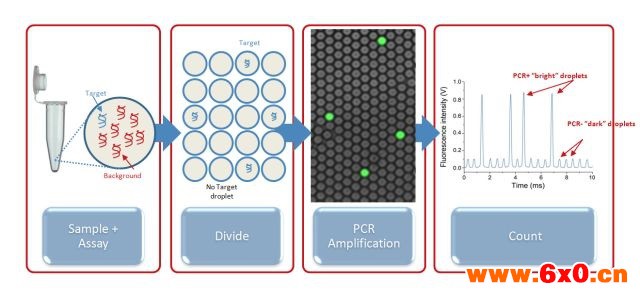
在实时定量 PCR(qPCR) 过程中,荧光信号随着扩增产物的积累而增强。qPCR 能够实时获得模板扩增的荧光值,然后根据DNA 模板在指数增长时期的 Ct 值与标准 DNA 的Ct值比较来计算初始模板的浓度。但是这种方法由于是大体积反应系统,非特异性的扩增增加了假阳性结果和背景信号,因此,最终无法获得定量的结果。
随着MEMS工艺的不断成熟和微流控技术的不断发展,新一代PCR技术,数字PCR(digital PCR, dPCR) 也随之出现。digital PCR是一种新的定量PCR,主要是对PCR反应物进行有限稀释,随后在不同的反应腔室里进行PCR扩增,最后根据泊松分布原理及阳性微滴的个数与比例得出靶分子的起始拷贝数或浓度的技术。
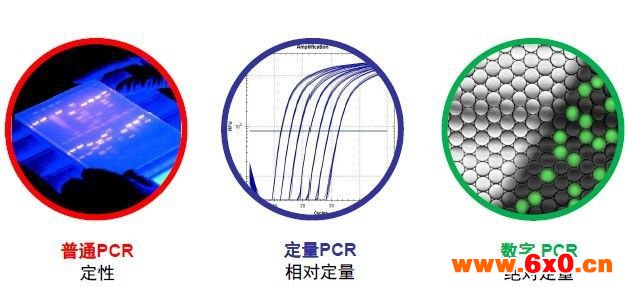
图1. PCR技术的发展历程
二. 数字微流控(dPCR)的原理
20 世纪末,Vogelstein 等提出数字PCR( digitalPCR,dPCR) 的概念,通过将一个样本分成几十到几万份,分配到不同的反应单元,每个单元包含一个或多个拷贝的目标分子( DNA 模板) ,在每个反应单元中分别对目标分子进行PCR 扩增,扩增结束后对各个反应单元的荧光信号进行统计学分析。
dPCR是一种核酸分子定量技术。
dPCR一般包括两部分内容,即PCR扩增和荧光定量分析。
在PCR 扩增阶段,与传统技术不同,dPCR一般需要将样品稀释到单分子水平,并平均分配到几万个反应腔室里反应。这样子相当于变相的对靶基因进行富集。与此同时,由于对原样品的大幅度的稀释,使得PCR抑制剂浓度显著降低,这样dPCR对初始PCR反应物里抑制剂的要求显著低于qPCR。
不同于 qPCR对每个循环进行实时荧光测定的方法,dPCR 技术是在扩增结束后对每个反应单元的荧光信号进行采集,最后根据泊松分布原理及阳性微滴的个数与比例得出靶分子的起始拷贝数或浓度。
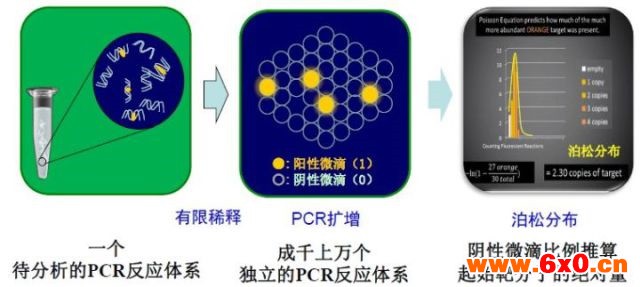
图2. dPCR基本原理
三. dPCR与qPCR的对比
相对于荧光定量PCR(qPCR)而言,dPCR具备以下优势:
1、 灵敏度可达单个核酸分子:检测限低至0.001%,原因在于dPCR可以实现靶标DNA/RNA的富集;
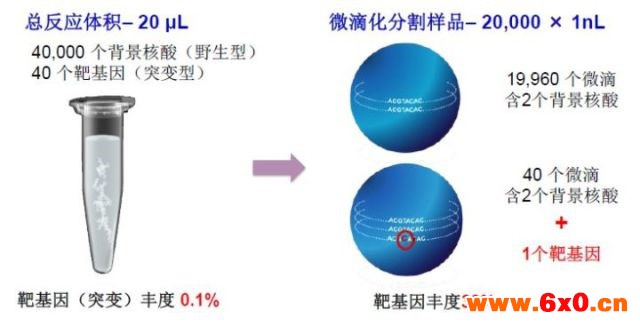
图3. dPCR可以实现痕量核酸的高灵敏检测
2、 无需标准品(标准曲线),即可对靶分子起始量进行定量;
3、 特别适合基质复杂样品的检测:终点PCR检测,不依赖Ct值,不依赖扩增效率,能克服PCR抑制剂的影响,适合动血样、FFPE组织、粪便、尿液、痰液、水样、土壤、植物等复杂样品中DNA的定量;
4、 能够有效区分浓度差异(变化)微小的样品:更好的准确度、精密度和重复性,可以用于测定靶基因的相对表达,基因拷贝数变异分析等。








 QQ交流群
QQ交流群

