

【英国杜伦大学案例】吲哚-3羧酸酯及其类植物生长素衍生物的流动合成
吲哚类化合物是色氨酸(1)、相关的神经递质血清素(2)以及许多复杂的生物碱天然物质和药物中常见的最重要的生物活性杂环结构之一。吲哚类植物激素在促进和调节植物生长发育方面也发挥着重要作用。的确,80%的植物自身合成的生长激素含有吲哚结构(图1,结构3-7)。由于它们的调节活动,这些结构已成为研究促进植物生长以及目标植物的生长抑制作用产生新型农用除草剂的主要目标。最近的一项合作研究了合成的吲哚-3-乙酸类似物在阔叶植物(双子叶植物)中的吸收及其分布,需要大量制备结构8(图1),以进行大规模的田间试验。此外,由于气候模式和环境问题对这类研究所需物质的时间安排和最终数量有重大影响,因此,人们还认为,能够使用灵活尺度的流动化学方法按需生产是非常可取的。
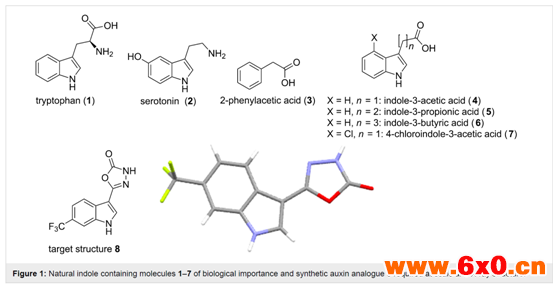
图1
自1835年费希尔首次合成吲哚类化合物的报告以来,又有十几种独特的吲哚类化合物被报道出来,显示出在这些有价值的结构中开发新化合物的重要性。然而,大多数吲哚类物质合成的共同特点是需要使用有害物质,如肼(Fischer)、重氮类化合物(Japp Klingemann)或叠氮化物(Hemetsberger Knittel),或者在吲哚环形成之前必须在多步反应中构建特定功能的前体。为了克服这些潜在的缺点,Marcus Baumann及其团队开始开发一种良性的工艺,依靠廉价的底物和无毒的试剂,用一种容易扩大且连续的方式迅速合成所需的吲哚。
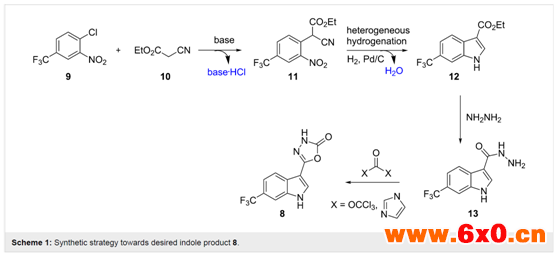
为了通过稳健的合成步骤生成核心吲哚单元,University of Durham化学系的Ian R. Baxendale教授团队决定研究以氰乙酸乙酯(10)为亲核试剂,在碱介导的SNAr反应中处理2-氯硝基苯9(Scheme1)。所得到的加合物11将在非均相氢化条件下,通过还原环化步骤生成吲哚产物12。从相应的酯功能化吲哚12,预期与肼缩合会得到相应的酰肼13,其可通过活性羰基供体的作用如CDI(1, 1-羰基二咪唑)或三光气(双(三氯甲基)碳酸酯)环构化得到所需的产品8。这种合成策略具有多种优势,因为它依赖于现成的底物和试剂,产生少量无毒副产物(碱?HCl、H2O),并在关键的环构化步骤中使用工业上较友好的加氢工艺。相关成果喜人,并于2017年发表在《BeilsteinJournal of Organic Chemistry》杂质上(Beilstein J. Org. Chem. 2017, 13, 2549–2560)。
为了开始这项研究,作者首先进行了一个全面的筛选流程,以确定形成化合物11的流动兼容性条件。对溶剂、碱、温度和试剂化学计量比的优化选择结果如表1所示。
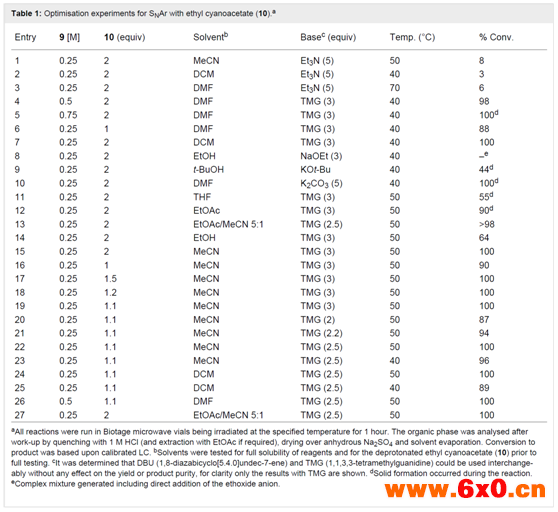
表1
虽然DMF和乙腈是很好的反应溶剂(表1,第3,6,10,15,23和26项),但在淬灭反应(1 M HCl)后有效地提取产品上遇到了困难。乙酸乙酯性能优异,可使萃取非常容易,但在反应过程中观察到少量暗红色且粘稠的沉淀物(表1,第12项)。在中试尺度的流动反应器工艺中这被认为是有问题的,因为可能造成堵塞。然而,研究发现,添加10~20% v/v的乙腈可以保证溶液完全均匀,也可以进行简单的水萃取,回收率较高(>90%)。然而,从初步结果来看,DCM是最有效的溶剂(表1,第7、24和25项),允许0.25 M的底物9的溶液在50℃时用TMG或DBU(2.5当量)和氰乙酸乙酯(10,1.1当量)进行定量转化。
基于这些筛选结果,作者设计了一个简单的流动装置,两股预备的原料液在一个简单的T型混合器中汇合,随后直接到一个维持在50℃的加热的流动线圈反应器中(Scheme 2)。迅速形成的深红色溶液(SNAr离子加合物)在保温周期(35 -108 min)后使用在混合后的混合物相分离之前,通过专用的混合芯片进行混合的第三股物料盐酸(1 M)淬灭。
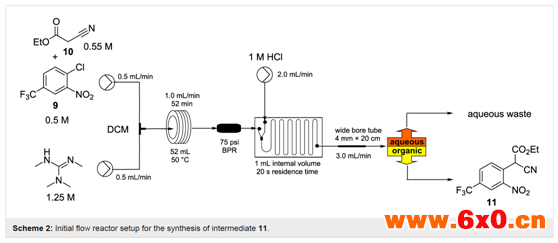
为了使相分离成为可能,作者使用了一个基于改进的Biotage通用分离器的被动膜系统。这使得较重的氯化物相可以从较低的连接处去除,较轻的水相可以从溢流位置流出。当从主反应器流出的流量为0.5-1.2 mL/min时,该装置可靠地进行了良好的淬灭和分离。然而,在高流速下产生了两相混合物的不完全分离(有些乳液形成),导致产品中的物质向水中流失从而造成损失,这是由于在较高的流速下在线搅拌芯片产生了较大的剪切力,以及形成高度柱塞流所需要的稳定时间更长。这个问题可以通过将水从第一个分离器引出到第二个等效装置,或者简单地通过两个平行分离器将原来的淬灭流股分开来解决。虽然这些方法在设计或使用分离器组件时不是优的,但在功能上是可行的。因此,作者还研究了用各种配置的T型和Y型连接器替换有问题的混合芯片,但这立即带来了其他问题,因为不完全淬灭导致了产品回收不良和相关的污染。一种更直接的方法是在分离器之前引入一个流动分层区,这是通过简便地引入一段内径更宽的管(从内径1.6 mm→4.0 mm×20 cm的反应器扩展而来)来实现的。这使得反应器可以通过2.0 mL的流量(对于1.0 mL以上的流量,则向系统中添加第二个52 mL的流动盘管)成功地在线淬灭反应,然后由单个Biotage通用分离器依次处理。在所有情况下,主反应器的出料都显示出定量转化,只有在溶剂蒸发后产品被分离出来。反应器流出速率为1.8 mL/min,这使得在稳态条件下的最大吞吐量达到27 mmol/h。
尽管该反应器功能多样,但其生产效率低于理想要求,不幸的是,DCM被证明对以下氢化步骤是一个不兼容的溶剂。虽然溶剂交换是可能的,但作者确定,当乙氧基乙酸作为溶剂并在乙酸作为添加剂存在下用乙醇稀释时,可使吲哚的还原环构化得以成功进行。在此基础上,研究了乙酸乙酯中中间体11的放大反应。在之前成功使用Coflore,AM技术公司ACR设备处理流动泥浆后,作者决定在合成中使用该设备来克服在溶解性方面遇到的问题。
起始物料用乙酸乙酯中分别储备的料液来制备,而后混合,首先是碱和氰乙酸乙酯(10)在芳氯9的溶液汇合前混合(注意这是一个放热过程),然后进入ACR,振动频率设置为8 Hz(Scheme 3)。
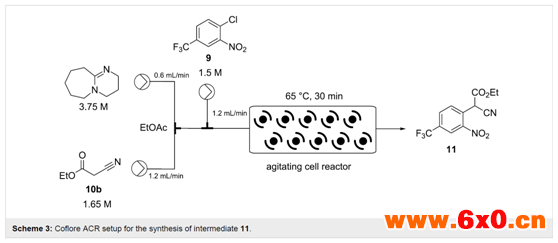
为了加强这一过程,并以减少溶剂量为具体目标,以便在随后的还原步骤(乙醇稀释后)中保持一个可行的反应浓度,作者进行了一系列浓度和流速优化。最终发现,在3 mL的混合流速下,相当于停留时间约为30 min,反应器温度为65℃,能够实现1.5 M底物9溶液的定量转化。连续运行时,反应器的出料为暗红色悬浮液,其中包括体积分数约为10%的固体,但即使在较长时间内(> 14 h)也能很容易地通过系统进行处理。该装置的理论工作效率为0.108 mol/h。接下来,为了便于整体淬灭和后处理,作者在以6 mL/min的流速输送的2 M HCl水溶液的汇合点添加了一个静态混合元件(Scheme 4)。通过将暗红色的反应混合物转化为收集时快速相分离的浅黄色双相溶液,可以立即观察到有效淬灭的迹象。通过对有机相的1H NMR和LC分析,确认了该方法淬灭成功,显示只有产物11和微量氰乙酸乙酯(10)。最后进行人工分离,然后在Na2SO4上干燥,将溶剂蒸发,得到了目标产物11,收率94-96%,基于每隔20 min取一次样,共取6次。
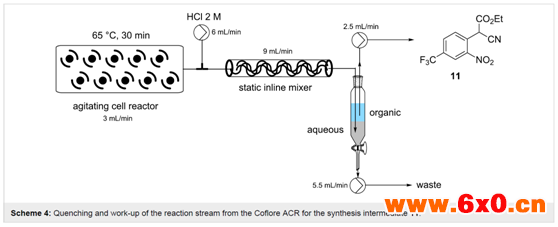
几种实现间歇分离自动化的实验室方法已经被报道,利用机器视觉系统和采用光学或感应电导率(阻抗测量)来确定相位划分的内联检测设备来确定相分离。然而,由于特定项目的时间限制,作者被迫使用更人工的方法,但肯定会将这种节省劳动力的设备纳入未来的发展计划。作者的基础系统使用一个简单的间歇式收集容器和两个位置合适的高效液相色谱泵来独立地去除水相和有机相。偶尔的人工干预允许间歇重新校准泵,以保持工作体积,并创建一个合适的相段进行去除。同样,在溶剂蒸发后,目标分子11的分离收率高达93-97%。
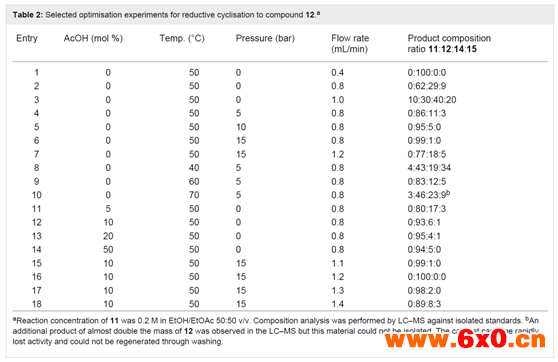
表2
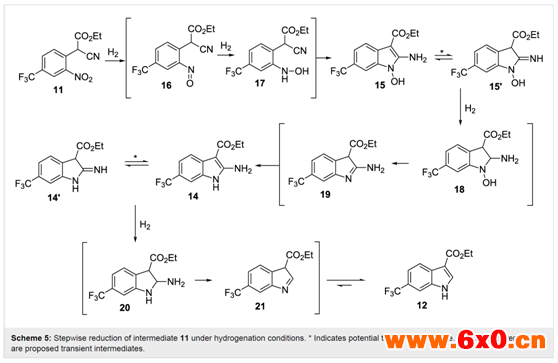
水相淬灭分离后收集的溶液中含有0.4 M产品11,该产品11再用乙醇稀释,以提供0.2 M的原液,用于下一个还原性成环步骤。在全氢条件下,使用含10 mol% Pd/C催化剂的ThalesNano H-cube系统进行还原。流速为0.4 mL/min,温度为50℃,完全转化为吲哚产物12(表2,第1项)。当流速提高到0.8-1 mL/min时,观察到原料完全消耗,但分离出混合产物(表2,第2、3项)。对其组成的详细分析表明了所需产物(12,62%)和另外两种化合物(图2)的存在,这两种化合物在柱层析分离后分别被确认为氨基吲哚(14,29%)和n-羟基氨基吲哚(15,9%)。正如预期,随着流速进一步增加,n-羟基氨基吲哚15的比例增加,后两种不完全还原产物增多。这与以往文献中使用催化氢化或化学计量金属(锌或铟)介导还原的研究结果相关联,并且与逐步还原机制相一致(Scheme 5)。从实验序列中可以明显看出,需要几倍当量的氢才能完全还原,因此较高的氢浓度(压力)是有益的(表2,第4-10项)。作者还注意到,根据机理,苯胺氮的质子化可以促进还原步骤,并有助于水(16和18)和氨(20)的去除。因此,作者筛选了一系列酸性催化剂,其中乙酸(10-30 mol%)(表2,第11-14项)。较强的酸或较高的酸负荷往往会导致产生较高的内部压力,在某些情况下,若观察到沉淀物的形成,则需要将运行提前终止并安全关闭。使用乙酸并限制酸的浓度(10 mol%),在较高的内部压力(15 bar)下运行,能够稳定且连续地操作,流量允许提高到1.3 mL/min,同时确保>98%的转化至所需的吲哚12(表2,第15-18项)。这转化为15.6 mmol/h(3.7 g/h产品)的通量。通过9:1正己烷/yi mi 溶剂蒸发和研磨,可分离出白色固体最终产物,收率93%。这种净化除去了残留的乙酸,杂质即使存在也只是痕量的副产物。
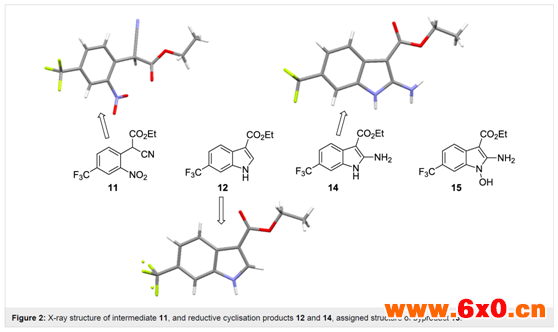
图2
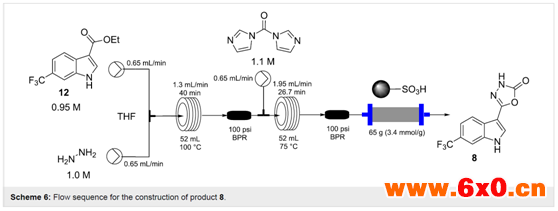
在接下来的实验序列中,作者研究了这个过程的最后两个步骤:乙氧基取代物的联氨,然后成环形成3H-[1, 3, 4]恶二唑-2-1单元。
在此过程中,将化合物12的0.95 M THF溶液与肼溶液(THF中,1.0 M)混合,直接进入加热的流动线圈中,在100℃下进行过热(Scheme 6)。停留时间为40 min,可以完全转换为相应的酰肼13,该酰肼13与含有CDI(THF中,1.1 M)的另一股料液直接混合,并在第二个流线圈中75℃加热26 min。这提供了最终产品8的定量转换,产品流通过QP-SA清除盒(一种磺酸功能化聚合物)易于提纯。该产品8经溶剂蒸发得到黄色固体(94%),但需要从DCM重结晶得到白色非晶粉末,纯度高,收率82%。
作者进行了一系列在最后一步中使用碳酸二甲酯作为CDI替代试剂的尝试,在一系列条件下都失败了,而在前面的酰肼形成步骤中直接使用(甲氧羰基)肼(CAS:6294-89-9)的试验也失败了。然而,作者发现三光气(0.4当量)可以成功地应用于后者的成环过程。使用与Scheme 6类似的反应器组件,将三光气(lv fang中,0.8 M)与中间体13的溶液混合,通过一个加热(55℃)的流动线圈,然后通过一个含有QP-DMA(N, N-二甲基苄胺聚苯乙烯)的填充床扫气筒。溶剂蒸发后,产品为白色固体,分离收率91%。在实践中,这一方法被证明远远优于以前使用的CDI环化直接从反应器获得更高的总收率和质量提升的产品。实际上,最终产品8的纯度足够直接应用,允许在反应装置中用<2 h从一个稳定的中间体12按需大规模生产。
总结:
1.按照文中所述的步骤,作者已经能够通过一个灵活的实验流程快速制备所需的目标分子8;
2. 在整个实验流程中,英国AM Technology公司的连续多级搅拌反应器Coflore ACR在第1步生成中间体11的反应中起到了关键作用,因为反应中会不断生成大量不溶的固体盐副产物,而Coflore ACR可以在防止堵塞的同时使反应连续稳定运行;
3. Coflore ACR反应器可以解决连续流反应中的多项问题,包括对反应的高效淬灭以及处理含不溶性固体的反应,体现了Coflore ACR反应器良好的实用性能;
4. 作者利用Coflore ACR反应器实现了第1步中间体11的连续化稳定生产,并与整个后续流程实现串联,表明不溶性固体不再是限制连续流生产的问题,该项技术可以在精细化工产品的大量制备中起到关键性作用;
5. Coflore ACR是已经成熟商业化的反应器,具备很好的规范性和通用性,对如含固等多相等复杂体系的工业化应用前景十分广阔。
参考文献:
doi:10.3762/bjoc.13.251
Beilstein J. Org. Chem. 2017, 13, 2549–2560.
公司简介:
深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司(连续多级搅拌反应器、催化加氢系统)、英国NiTech公司(连续结晶仪、连续合成仪)在中国区的代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。
公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。
公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及技术服务方案。








 QQ交流群
QQ交流群

